
Содержание
- О выборочном контроле качества лекарственных средств и их уничтожении
- Каким образом осуществляется проведение административной процедуры по выборочному контролю контролирующим органом?
- В каких случаях проводится повторный выборочный контроль качества лекарственных средств?
- Каков механизм изъятия недоброкачественных лекарственных средств из обращения?
- Кто имеет право заниматься уничтожением лекарственных средств, не прошедших контроль качества?
- Представление сведений в Росздравнадзор
- Защита прав субъектов
- Последствия несоответствия
- КОНТРОЛЬ КАЧЕСТВА ЛЕКАРСТВЕННЫХ СРЕДСТВ ПРОМЫШЛЕННОГО ПРОИЗВОДСТВА
О выборочном контроле качества лекарственных средств и их уничтожении
В соответствии со ст. 9 Закона N 61-ФЗ <1> государственный контроль обращения лекарственных средств включает в себя процедуру проведения контроля качества лекарственных средств при их гражданском обороте в форме выборочного контроля.
<1> Федеральный закон от 12.04.2010 N 61-ФЗ «Об обращении лекарственных средств».
Что такое выборочный контроль и в каких случаях он проводится, расскажем в статье.
В силу п. 1.2 Административного регламента <2> организация проведения экспертизы качества, эффективности и безопасности лекарственных средств представляет собой функцию Росздравнадзора по привлечению научных, иных организаций, ученых и специалистов для проработки вопросов качества, эффективности и безопасности лекарственных средств посредством проведения исследований, анализа и оценки объектов экспертизы, подготовки заключений относительно этих объектов. При возникновении необходимости в проведении данной экспертизы она осуществляется в соответствии с законодательством РФ.
<2> Административный регламент Федеральной службы по надзору в сфере здравоохранения и социального развития по исполнению государственной функции по организации проведения экспертизы качества, эффективности и безопасности лекарственных средств, утв. Приказом Минздравсоцразвития России от 30.10.2006 N 734.
Экспертиза качества, эффективности и безопасности лекарственных средств включает в себя такие административные процедуры, как (п. 1.13 Административного регламента):
- организация проведения экспертизы качества, эффективности и безопасности, осуществляемой в ходе рассмотрения документов и принятия решения о государственной регистрации лекарственного средства (при регистрации лекарственных средств);
- осуществление сбора и анализа информации о побочных эффектах применения лекарственных средств;
- организация проведения экспертизы качества при осуществлении предварительного государственного контроля лекарственных средств;
- организация проведения экспертизы качества при осуществлении выборочного государственного контроля лекарственных средств;
- организация проведения экспертизы качества при осуществлении повторного выборочного государственного контроля лекарственных средств;
- осуществление сбора и анализа информации о качестве лекарственных средств.
Мы остановимся только на двух из перечисленных выше административных процедур — выборочном контроле качества и безопасности лекарственных средств и повторном его проведении.
Каким образом осуществляется проведение административной процедуры по выборочному контролю контролирующим органом?
В силу пп. 4 п. 1.13 Административного регламента организация проведения экспертизы качества лекарственных средств в форме выборочного государственного контроля осуществляется на основании задания Росздравнадзора, формируемого по результатам сбора и анализа информации о качестве лекарственных средств.
В целях повышения эффективности мероприятий по проведению контрольных процедур Росздравнадзор ежегодно информирует отечественные организации-производители, зарубежные компании — производители и организации-импортеры лекарственных средств о перечне лекарственных средств, подлежащих выборочному государственному контролю. Согласно Письму Росздравнадзора от 30.11.2010 N 04И-1208/10 в перечень лекарственных средств, подлежащих выборочному контролю качества в 2011 г., попали:
- фармацевтические субстанции;
- лекарственные препараты для лечения заболеваний желудочно-кишечного тракта;
- нестероидные противовоспалительные препараты;
- лекарственные препараты для лечения органов дыхания;
- антибиотики, противомикробные препараты и антисептические лекарственные средства;
- лекарственные препараты для лечения сердечно-сосудистых заболеваний;
- препараты, получаемые из крови и плазмы донорской;
- лекарственные средства для парентерального питания, инфузионные растворы и кровезаменители;
- лекарственные препараты, по которым в 2010 г. выявлены факты несоответствия требованиям нормативной документации;
- лекарственные препараты, по которым в 2009 — 2010 гг. выявлены факты фальсификации;
- впервые ввозимые и впервые производимые лекарственные средства;
- лекарственные средства, имеющие спектры сравнения для определения подлинности неразрушающими экспресс-методами.
В силу п. 1.5 Административного регламента Росздравнадзор на договорной основе вправе привлекать для проведения экспертизы качества лекарственных средств независимые экспертные организации. В качестве экспертных организаций могут выступать центры контроля качества (контрольно-аналитические лаборатории) субъектов РФ, аккредитованные Ростехрегулированием в качестве испытательных лабораторий.
Проведение экспертизы контроля качества и безопасности лекарственных средств осуществляется в форме заданий, которые могут быть общими и частными. Общее задание выдается экспертной организации на определенный период времени и устанавливает ее полномочия по проведению определенного вида экспертизы в течение указанного срока. Частное задание выдается экспертной организации на разовое проведение конкретного вида экспертизы. Задания на проведение экспертизы утверждаются руководителем Росздравнадзора или должностным лицом, специально уполномоченным для этой цели (п. 1.7 Административного регламента).
Отбор образцов лекарственных средств и передача их в экспертную организацию производятся в течение пяти рабочих дней с даты утверждения задания на проведение экспертизы (п. 3.5.2 Административного регламента).
В рамках инспекционного контроля за обращением сертифицированных лекарственных средств при поступлении их на территорию субъекта РФ осуществляется экспертиза качества лекарственных средств по трем показателям («Описание», «Упаковка», «Маркировка»), проверяются происхождение, соответствие лекарственного средства сопроводительным документам и государственному стандарту качества. В случае сомнения в достоверности полученных данных проводятся дополнительные испытания качества.
Результаты проведенных испытаний, информация о выявлении в обращении недоброкачественных и фальсифицированных лекарственных средств направляются в режиме on-line от центров контроля качества в Росздравнадзор для принятия решения о возможности дальнейшего обращения этих препаратов.
Срок для принятия решения в отношении лекарственных средств, проходящих контроль качества, — пять рабочих дней с даты получения результатов экспертизы (п. 3.5.5 Административного регламента).
В каких случаях проводится повторный выборочный контроль качества лекарственных средств?
В соответствии с п. 3.6 Административного регламента административная процедура по проведению экспертизы качества повторного выборочного государственного контроля лекарственных средств выполняется в случаях возникновения сомнений в их качестве у субъекта обращения лекарственных средств в связи с поступлением от него комплекта соответствующих документов и данных.
Поступившие документы и данные, содержащие сведения, дающие основания для сомнений в качестве лекарственного средства и предназначенные для проведения экспертизы качества в виде повторного выборочного государственного контроля лекарственных средств, поступившие от организации, регистрируются в течение одного рабочего дня с даты их получения.
Росздравнадзор в течение пяти рабочих дней проверяет поступившие документы на обоснованность проведения экспертизы качества. Надлежащим основанием считается выявление несоответствия показателей качества лекарственного средства требованиям государственного стандарта качества (нормативной документации) и несогласие с этим организации-производителя или импортера.
При отсутствии надлежащих оснований Росздравнадзор готовит письменный отказ в проведении экспертизы качества при осуществлении повторного выборочного государственного контроля лекарственных средств с указанием оснований отказа, который подписывается его руководителем и направляется в обратившуюся организацию.
Если основания обоснованны, на основании полученного от Росздравнадзора задания экспертная организация проводит экспертизу качества образцов в срок, определяемый заданием, но не превышающий 30 дней с даты получения образцов экспертной организацией. Ответственность за контроль хода проведения экспертизы несет начальник отдела, осуществляющего контроль производства лекарственных средств.
Каков механизм изъятия недоброкачественных лекарственных средств из обращения?
В соответствии с п. 3 Правил уничтожения <3> Росздравнадзор в случае выявления фактов ввоза на территорию РФ или обращения на территории РФ недоброкачественных и (или) фальсифицированных лекарственных средств принимает решение, обязывающее владельца указанных лекарственных средств осуществить их изъятие, уничтожение и вывоз в полном объеме с территории РФ. Решение должно содержать:
- сведения о лекарственных средствах;
- основания для изъятия и уничтожения лекарственных средств;
- срок изъятия и уничтожения лекарственных средств;
- сведения о владельце лекарственных средств;
- сведения о производителе лекарственных средств.
<3> Правила уничтожения недоброкачественных лекарственных средств, фальсифицированных лекарственных средств и контрафактных лекарственных средств, утв. Постановлением Правительства РФ от 03.09.2010 N 674.
Владелец недоброкачественных и (или) фальсифицированных лекарственных средств в срок, не превышающий 30 дней со дня вынесения Росздравнадзором решения об их изъятии, уничтожении и вывозе, обязан исполнить это решение или сообщить о своем несогласии с ним. В случае если он не согласен с вынесенным решением, а также если он не выполнил это решение и не сообщил о принятых мерах, Росздравнадзор обращается в суд. В дальнейшем спорную ситуацию разрешает суд. Он выносит решение, касающееся изъятия и уничтожения недоброкачественных и (или) фальсифицированных лекарственных средств (п. п. 4, 5 и 7 Правил уничтожения).
Кто имеет право заниматься уничтожением лекарственных средств, не прошедших контроль качества?
Согласно п. 6 ст. 59 Закона N 61-ФЗ и п. 8 Правил уничтожения уничтожение недоброкачественных лекарственных средств осуществляется организацией, имеющей лицензию на деятельность по сбору, использованию, обезвреживанию, транспортировке и размещению отходов I — IV класса опасности, на специально оборудованных площадках, полигонах и в специально оборудованных помещениях с соблюдением требований по охране окружающей среды в соответствии с законодательством РФ.
Расходы, связанные с уничтожением недоброкачественных лекарственных средств, возмещаются их владельцем. Владелец недоброкачественных лекарственных средств, принявший решение об их изъятии, уничтожении и вывозе, передает их организации, осуществляющей уничтожение лекарственных средств, на основании соответствующего договора (п. п. 9 — 10 Правил уничтожения).
В день уничтожения недоброкачественных лекарственных средств организация, осуществляющая это, составляет соответствующий акт (п. п. 11, 12 Правил уничтожения).
М.Р.Зарипова
Эксперт журнала
«Аптека: бухгалтерский учет
и налогообложение»
Представление сведений в Росздравнадзор
Обязанность по предоставлению информации о сериях (партиях) поступающих в гражданский оборот, возложена на производителей лекарственных средств, осуществляющих производство в России, и на организации, которые ввозят лекарственные средства в Россию. Сведения, перечень которых определён в Приказе, подаются через подсистему «Выборочный контроль» автоматизированной системы Росдравнадзора. На сайте Росздравнадзора сейчас размещена информация о порядке получения/переполучения доступа в указанную систему.
Срок для предоставления информации установлен в 5 рабочих дней с момента поступления лекарственного средства в гражданский оборот. Для производителей точкой отсчёта является разрешение на выпуск, т.е. подтверждение уполномоченным лицом производителя соответствия серии лекарственного средства требованиям, установленным при их государственной регистрации. Для организации, осуществляющей ввоз лекарственных средств, – выпуск таможенными органами лекарственных средств под определенную таможенную процедуру. В случае ввоза лекарственных средств в Российскую Федерацию с территории государств-членов Евразийского экономического союза – с даты осуществления ввоза лекарственных средств на территорию Российской Федерации.
Полученные в ходе сбора информации сведения в дальнейшем используются Росздравнадзором для формирование плана осуществления выборочного контроля качества лекарственных средств.
При формировании плана Росздравнадзором учитываются следующие критерии: результаты проверок, проводящихся в рамках государственного контроля за обращением лекарственных средств, результаты выборочного контроля в предыдущие периоды, сведения о выявленных фальсифицированных или недоброкачественных лекарственных средствах, обращения граждан, юридических лиц, сведения из СМИ. Иными словами, выборочный контроль добросовестных участников обращения лекарственных средств будет осуществляться реже. В то же время необходимо подчеркнуть, что, как именно каждый из критериев влияет на интенсивность выборочного контроля, Приказ не регулирует.
Отметим, что положения о предоставлении информации вступают в силу с 14 января 2016 года.
Защита прав субъектов
Безусловным преимуществом Приказа является урегулированность самой процедуры отбора лекарственных средств, что является одной из гарантий защиты прав и законных интересов субъектов обращения лекарственных средств. Так, любой отбор образцом должен быть подтверждён целым рядом документов.
План выборочного контроля. Формируется до 1 января каждого календарного года и содержит перечень лекарственных средств, количество испытаний лекарственных средств на год. План утверждается руководителем Росздравнадзора и публикуется на официальном сайте ведомства.
Задания Росздравнадзора. Формируются на основе плана или в связи с поступлением информации об угрозе причинения вреда или причинении вреда жизни и здоровью граждан. Могут быть утверждены руководителем Росздравнадзора или его заместителями. В задании должен быть определён объект испытаний, виды и методы испытаний, их срок и особенности.
Требования о предоставлении образцов лекарственных средств. На их основании производится непосредственно отбор образцов. Они включают в себя ссылку на задание Росздравнадзора, наименование проверяемой организации, перечень требуемых образцов. Представитель организации в обязательном порядке должен быть ознакомлен с требованием, о чём в нём делается соответствующая отметка.
Протоколы отбора образцов. В них фиксируется состав комиссии, осуществляющей отбор, список отобранных образцов, условия хранения на момент отбора и результаты внешнего осмотра лекарственных средств. Протоколы, соответственно, оформляются в двух экземплярах.
Последствия несоответствия
В случае, если в ходе выборочного контроля будет выявлено несоответствие лекарственного средства обязательным требованиям, может быть принято решение о его переводе на посерийный контроль. При этом расходы, связанные с таким переводом, возлагаются на производителя лекарственных средств либо на владельца регистрационного удостоверения.
В случае невыполнения предписания, выданного по итогам выборочного контроля, может быть рассмотрен вопрос о приостановлении применения лекарственного препарата.
Подводя итог, отметим, что цель выборочного контроля – обеспечить качество и безопасность лекарственных средств, обращающихся на территории Российской Федерации. При этом выборочный контроль не должен стать непосильной нагрузкой для добросовестных участников фармацевтического рынка, интересы которых учтены и защищаются Приказом.
Все без исключения лекарственные препараты, допущенные к продаже через аптечную сеть, проходят обязательный контроль качества. О том, что представляет собой система контроля качества лекарственных средств, и пойдет речь. Но прежде, чем начать разговор, необходимо уточнить, что следует понимать под «качеством» того или иного лекарства.
Ни в коем случае не следует путать «эффективность» медикамента с его «качеством». Дело в том, что ожидаемый эффект от конкретного лекарственного препарата зависит от множества факторов, в том числе и от условий его приема, функционирования органов пищеварения и выделения скорости метаболизма в клетках печени. Что же понимают под качеством лекарственного средства?
На что следует обращать внимание при проведении контроля качества лекарственных препаратов?
Признак качества №1 – химическая чистота.
Обращают внимание на процентное содержание посторонних примесей и их состав. Лекарственное вещество не должно содержать даже незначительное количество вредных для здоровья веществ.
Признак качества №2 – точное соответствие химического состава.
Каждая лекарственная форма должна содержать точное количество заявленного действующего вещества. Контроль качества лекарственных средств включает не только исследование на точное химическое соответствие тому, что заявлено, но и определение количества действующего вещества в каждой ампуле или таблетке.
Признак качества №3 – безопасность всех ингредиентов.
Многие лекарственные формы, к числу которых относятся таблетки, свечи, сиропы, микстуры, имеют сложный состав. В ходе контроля качества лекарственных препаратов проводится тщательное исследование всех дополнительных ингредиентов.
Признак качества №4 – качество упаковки.
От качества упаковки зависит не только удобство в процессе транспортировки, хранения, продажи и использования медикаментов, но и срок годности.
Контроль качества лекарственных средств включает в себя процедуры по отбору проб, проведению органолептических, гигиенических, химических, микробиологических, радиологических и иных исследований. Главная цель всей сложной системы контроля качества состоит в недопущении к использованию медикаментов, не соответствующих утвержденным стандартам.
В компании «КоролёвФарм» постоянно проводится контроль качества медицинской и косметической продукции в точном соответствии с ныне действующими нормативными документами и международными стандартами.
Организация контроля качества лекарственных средств – это многоступенчатый процесс, который начинается с контроля качества сырья и заканчивается анализом готовой продукции. В компании «КоролёвФарм» , как и на каждом фармацевтическом предприятии, имеется свой собственный центр контроля качества лекарственных средств. Данная служба занимается решением организационно-методических вопросов, контролем всех этапов производства, исследованием готовой продукции.
Основные ступени контроля качества лекарств и косметологической продукции в компании «КоролёвФарм»:
Ступень №1 — не только контроль качества сырья и готовой продукции, но и все этапы производства выполняются квалифицированным персоналом.
Ступень №2 — каждый отдельный этап производства регламентируется соответствующими инструкциями и по мере надобности протоколируется.
Ступень №3 — обработка и хранение сырья, промежуточной и готовой продукции осуществляется в строгом соответствии с установленными стандартами.
Ступень №4 — соблюдение мер по предупреждению микробного, радиационного и других видов загрязнения на всех этапах производства и хранения.
Ступень №5 — качество готовой продукции обеспечивается наличием современного высокоточного оборудования.
Ступень №6 — качество готовой продукции во многом определяется качеством сырья. На сотрудников центра контроля качества лекарственных средств возлагаются обязанности по недопущению в производство сырья, не соответствующего установленным требованиям, в том числе и с истекшим сроком годности.
Ступень №7 — упаковка готовой продукции проводится в условиях, исключающих возможность перепутывания, подмены или загрязнения. Вся готовая продукция маркируется в процессе упаковки, то есть немедленно.
Контроль качества лекарств и косметики в «КоролёвФарм» проводится, как сотрудниками компании, так и независимыми контролерами, в том числе и аудиторами. Контроль качества лекарственных средств регламентируется приказами Минздрава РФ и другими нормативными документами.
КОНТРОЛЬ КАЧЕСТВА ЛЕКАРСТВЕННЫХ СРЕДСТВ ПРОМЫШЛЕННОГО ПРОИЗВОДСТВА
Производство ЛС — это серийное получение ЛС в соответствии с правилами организации производства и контроля их качества, утвержденными соответствующим федеральным органом.
Производство ЛС осуществляется предприятиями-производителями, имеющими лицензии на их производство. Государственный контроль производства выполняют федеральный и территориальный органы контроля качества ЛС, в права которых входит:
— беспрепятственный доступ на любое предприятие-производитель ЛС и контрольное изъятие производимых образцов;
— снятие копий с документов, необходимых для проведения контроля производства и качества ЛС;
— запрет производства и продажи уже произведенных ЛС в случаях: 1) если ЛС не прошли государственную регистрацию в РФ (исключение — ЛС, предназначенные для проведения клинических исследований); 2) отсутствия лицензии на производство; 3) изготовления с нарушением правил организации производства и контроля качества ЛС (статья 13 Государственного Закона о лекарственных средствах).
Лицензия на производство ЛС выдается федеральным органом исполнительной власти на срок не менее 5 лет. В случае, если предприятием-производителем изменены условия производства, оно обязано получить новую лицензию на производство.
На фармацевтическом предприятии контроль качества ЛС является частью надлежащей производственной практики, гарантирующей правильность процедуры отбора проб, подготовки пробы к анализу (пробоподготовки), определения действующего вещества, принятия решения о приеме испытуемого материала в соответствии с требованиями, установленными в НД (ФСП, спецификации).
Отдел контроля качества (ОКК) фармацевтического предприятия осуществляет различные типы фармацевтического контроля: входной контроль фармацевтических субстанций и вспомогательных веществ, межоперационный контроль в процессе производства и контроль качества готовой продукции (рис. 6.2).
Таким образом, контроль качества на производстве включает контроль исходного сырья, полупродуктов, лекарственных субстанций и готовых лекарственных форм.
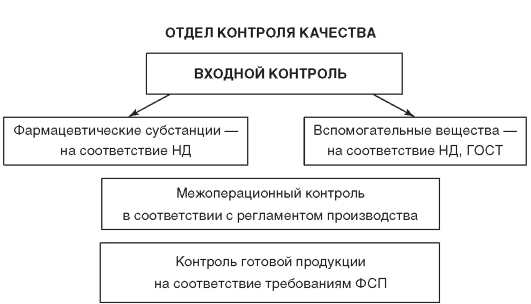
Рис. 6.2. Типы фармацевтического анализа в условиях производства лекарственных средств
(по:Елизарова Т.Е. Современные методы стандартизации и контроля качества лекарственных средств. — М.: МИА, 2008)
В период бурного развития фармацевтической промышленности возникли проблемы качества ЛС, которые не могли быть решены только путем усиления фармакопейного анализа. Обеспечение качества ЛС стало возможным только на базе проведения фармацевтического анализа в соответствии с правилами GMP.
Согласно правилам GMP, объектом контроля становится весь процесс производства ЛС, включая помещения, персонал, документацию. В России правила GMP нашли отражение в виде ГОСТ Р 52249-2009 «Правила производства и контроля качества лекарственных средств».
Стандарт содержит требования к производству и контролю качества ЛС для человека и животных. Стандарт распространяется на все виды ЛС и устанавливает общие требования к их производству и контролю качества, а также специальные требования к производству отдельных видов ЛС. Стандарт не распространяется на обеспечение промышленной безопасности, пожарной безопасности, взрывобезопасности, химической безопасности и безопасности других видов при производстве ЛС, требования к которым приведены в других НД.
Надлежащая практика контроля качества фармацевтических препаратов обеспечивается комплексом мероприятий при их разработке и исследовании с учетом требований GMP. Каждая методика
должна содержать обоснование преимуществ по сравнению с другими в виде представленных результатов сопоставления ее применения (валидация).
Валидация методавключает следующие метрологические характеристики:
— правильность (accuracy) — близость результатов к истинному значению, что может быть проведено при сравнении с результатами, полученными с помощью иной методики, валидированной ранее;
— точность (precision) — согласованность между отдельными результатами испытаний (отклонение отдельных результатов от среднего значения — относительное стандартное отклонение);
— сходимость (repeatability) — точность методики при ее выполнении одним и тем же аналитиком при одних и тех же условиях (реактивы, оборудование, лаборатория);
— воспроизводимость (reproducibility) — точность методики при использовании ее в различных условиях для идентичных образцов, отобранных из одной и той же однородной серии материала (разные лаборатории, исполнители, оборудование, время);
— надежность (robustness) — способность методики давать результаты анализа с приемлемой правильностью и точностью при изменении условий работы для предположительно идентичных образцов из одной и той же однородной серии материала;
— чувствительность (sensitivity) — способность методики испытания регистрировать небольшие изменения концентрации (наклон калибровочной кривой);
— предел обнаружения (limit of detection) — наименьшее содержание, при котором анализируемое вещество может быть обнаружено.
Контроль качества ЛС на отдельных технологических стадиях его получения обеспечивает надлежащее качество конечного продукта.
Для лекарственных препаратов регламентируется надлежащая микробиологическая чистота. Загрязнение микроорганизмами может происходить на разных стадиях производства, поэтому испытания на микробиологическую чистоту проводят на всех стадиях получения ЛС. Основными источниками микробной контаминации являются сырье, вода, оборудование, воздух производственных помещений, упаковка готовой продукции, персонал.
Для количественного определения содержания микроорганизмов в воздухе используют различные методы отбора проб: фильтрацию, осаждение в жидкостях, осаждение на твердые среды. Для оценки микробиологической чистоты выполняют тесты на стерильность.
При определении стерильности ЛС, обладающих выраженным антибактериальным действием, бактериостатическими, фунгистатическими свойствами, а также содержащих консерванты, используют метод мембранной фильтрации.
Метод мембранной фильтрации основан на пропускании ЛС через полимерную мембрану. При этом микроорганизмы остаются на поверхности мембраны. Затем мембрану помещают в соответствующую питательную среду и наблюдают образование колоний при инкубировании.
Также проводят испытания на пирогенность инъекционных препаратов.Необходимость проведения этого теста связана с присутствием в инъекционных препаратах фрагментов клеток грамположительных и грамотрицательных бактерий, грибов, вирусов и эндотоксинов. Применение таких препаратов вызывает жар, озноб, тошноту, иногда летальный исход. Наибольшую опасность представляют эндотоксины, которые являются термостабильными и состоят из липополисахаридов внешней плазматической мембраны грамотрицательных бактерий, попадание которых в организм человека провоцирует резкий воспалительный процесс.
Контроль содержания пирогенных примесей в инъекционных препаратах проводят двумя методами: в опытах in vivo на кроликах и in vitro, с использованием ЛАЛ-реактивов, приготовленных из водного экстракта (лизата) амебоцитов мечехвоста полифема1.
ЛАЛ-тест впервые был включен в Фармакопею США в 1980 г., а позже был признан и в европейских странах. В 1997 г. в РФ была утверждена ФС «Определение содержания бактериальных эндотоксинов (ЛАЛ-тест)».
Основными преимуществами ЛАЛ-теста по сравнению с традиционным испытанием на кроликах является возможность оценки уровня бактериальных эндотоксинов в тех препаратах, которые нельзя испытывать на животных; более высокая (в 100 раз) чувствительность ЛАЛ-теста; быстрота выполнения (одно испытание занимает около 1,5 ч); испытания проводит один человек.
Более высокая чувствительность и быстрота ЛАЛ-теста обеспечивают возможность контроля качества воды для приготовления инъекционных ЛС в условиях производства. Этот тест используется для анализа «Воды инъекционной в ампулах», «Раствора натрия хлорида 0,9% для инъекций», различных лекарственных форм инсулина.
Под чувствительностью ЛАЛ-реактива понимают минимальную концентрацию международного.стандарта эндотоксина, которая приводит к образованию плотного геля при реакции с данным ЛАЛ-реактивом в гель-тромб тесте, проведенном при стандартных условиях.
Испытания на кроликах проводят на 12 кроликах породы шиншилла массой 2,5-3,0 кг с соблюдением строгого стандартного рациона питания в специально оборудованном тихом, светлом, без перепадов температуры помещении. Кроликов помещают в определенным образом оборудованные индивидуальные боксы, позволяющие проводить постоянное измерение температуры, которая записывается в автоматическом режиме самописцем, что исключает фальсификацию данных.
В начале опыта препарат вводят 3 животным. ЛС считается апирогенным, если суммарное повышение температуры тела животных (∑Δt) составляет не более 1,2 °С. На втором этапе испытаний препарат вводят 6 животным. ЛС признают пирогенным, если ∑Δt>3,0 °С. На третьем этапе число животных равно 9. ЛС считают пирогенным при ∑Δt>4,5 °С. Наконец, на четвертом этапе используется 12 животных, и препарат считают пирогенным, если ∑Δt>5,4 °С. После окончания проверки составляют протокол, который включает параметры эксперимента, заключение о соответствии проверяемого раствора требованиям на пирогенность.
Метод имеет ряд недостатков. В эксперименте используют животных, чувствительность которых к пирогенам в 3-4 раза ниже, чем у человека. Это требует соответствующего увеличения тест-дозы. Кроме того, многие ЛВ в дозах, близких к терапевтическим, могут вызвать токсические реакции и даже гибель животных,
поэтому используется заниженная величина тест-доз (инфузионные растворы глюкозы, антибиотики — бензилпенициллина натриевая соль, линкомицина гидрохлорид и др.). На проведение одной серии опыта требуется около 5 ч.
1 Мечехвост полифем, краб-подкова, королевский краб. Вид: Limulus polyphemus (Linnaeus, 1758).
LAL(ЛАЛ)-тест (Limulus Amebocyte Lysate) — ферментативная реакция ЛАЛреактива с эндотоксином, в результате которой образуется плотный гель. При отсутствии эндотоксина в анализируемой пробе гель не образуется. Положительный результат ЛАЛ-теста указывает на то, что в исследуемой пробе содержание эндотоксина составляет не менее 0,25 ЕЭ (единиц эндотоксина). Отрицательный результат говорит о том, что в исследуемой пробе содержание эндотоксина — менее
0,25 ЕЭ.
Контрольные вопросы и задания
• Охарактеризуйте этапы фармацевтического анализа: определение подлинности, оценка чистоты (определение примесей), количественный анализ.
• Перечислите правила надлежащей деятельности (GP), в соответствии с которыми должны осуществляться производство и контроль качества ЛС.
• Каковы особенности фармацевтического анализа воспроизведенных ЛС?
• Перечислите типы эквивалентности ЛС. Как производят оценку фармацевтической и биологической эквивалентности?
• Объясните необходимость применения в фармацевтическом анализе стандартных образцов сравнения.
• Перечислите характеристики валидации аналитического метода.